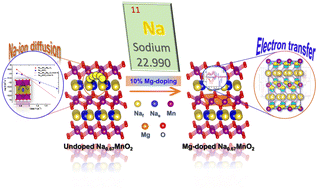
Research: Computational and Theoretical Chemistry
|
- For prospective research students, who wish to join our group - If you have CSIR/UGC-JRF fellowship; you can join the group through out the year.
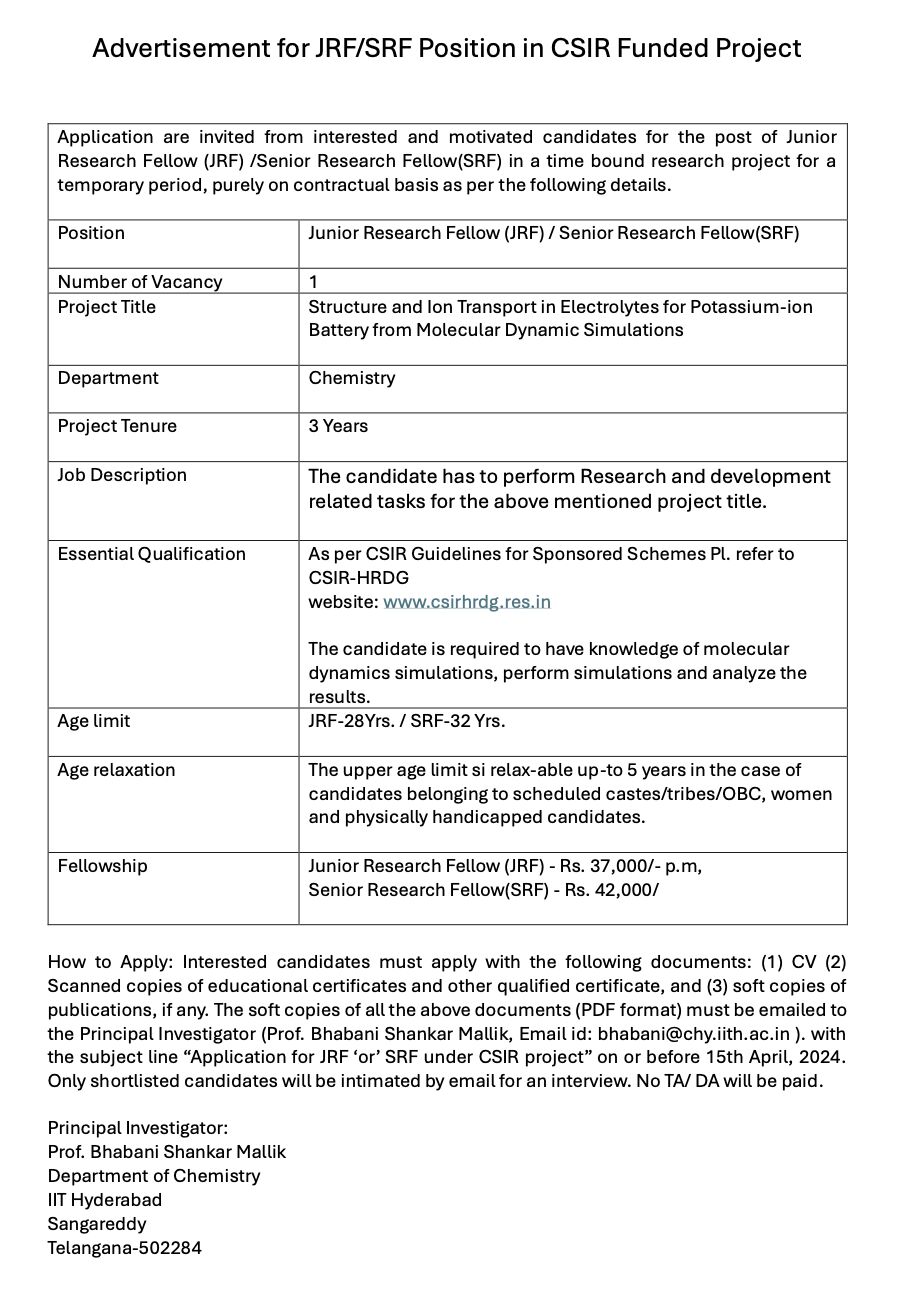
Advertisement of JRF/SRF position in a CSIR funded project at IIT Hyderabad. Please send the CV along with other documents to bhabani@chy.iith.ac.in
|
Teaching:
- CY 8xxx: Modern simulation methods
- CY 6903: Molecular modelling of complex chemical systems
- CY 6220: Physical methods in chemistry
- CY 6230: Principles of quantum chemistry
- CY 5250: Chemical binding and Molecular symmetry
- CY 5240: Quantum chemistry and molecular spectroscopy
- CY 1031: Dynamics of chemical systems (2 credits)
- CY 1020: Dynamics of chemical systems (1 credit)
- IC 1050: Introduction to quantum chemistry (2 credit)
Scientific Society
- Life Member (2785), Chemical Research Society of India (CRSI)
- Patron Member (PM/197/21), Orissa Chemical Society (OCS)
|